
La electroforesis es una técnica para la separación de moléculas según la movilidad de estas en un campo eléctrico. La separación puede realizarse sobre la superficie hidratada de un soporte sólido (p. ej., electroforesis en papel o en acetato de celulosa), o bien a través de una matriz porosa (electroforesis en gel), o bien en disolución (electroforesis libre). Dependiendo de la técnica que se use, la separación obedece en distinta medida a la carga eléctrica de las moléculas y a su masa.
La variante de uso más común para el análisis de mezclas de proteínas o de ácidos nucleicos utiliza como soporte un gel, habitualmente de agarosa o de poliacrilamida. Los ácidos nucleicos ya disponen de una carga eléctrica negativa, que los dirigirá al polo positivo, mientras que las proteínas se cargan al unirse con sustancias como el SDS (detergente) que incorpora cargas negativas de una manera dependiente de la masa molecular de la proteína. Al poner la mezcla de moléculas y aplicar un campo eléctrico, éstas se moverán y deberán ir pasando por la malla del gel (una red tridimensional de fibras cruzadas), por lo que las pequeñas se moverán mejor, más rápidamente. Así, las más pequeñas avanzarán más y las más grandes quedarán cerca del lugar de partida.
Electroforesis
La gran mayoría de macromoléculas están cargadas eléctricamente y, al igual que los electrolitos, se pueden clasificar en fuertes y débiles dependiendo de la constante de ionización de grupos ácidos y básicos. Por ejemplo los ácidos nucleicos son poliácidos fuertes.Por lo general, para caracterizar la molécula se determina la velocidad a la que esta se mueve en un campo eléctrico y se utiliza para determinar, en el caso de proteínas, la masa molecular o para detectar cambios de aminoácidos y separar cuantitativamente distintas especies moleculares; en el caso de ácidos nucleicos se determina su tamaño, medido en pares de bases
Velocidad de una molécula
Para separar distintas especies moleculares, se crea un campo eléctrico para la molécula colocada en un líquido portador. Al generar este campo existirá una intensidad pasando constantemente del polo positivo al polo negativo, por lo tanto, actuará una fuerza sobre la molécula y esta experimentará una aceleración hasta obtener una velocidad en la que la resistencia, por viscosidad del medio, neutraliza la fuerza impulsora, es decir, la molécula se desplaza con una velocidad constante:
Se asume que la partícula es esférica y a partir de la Ley de Stokes se obtiene
que

Por lo tanto la velocidad será:

Factores que afectan a la electroforesis
En general la electroforesis depende directamente del campo eléctrico y este
depende de distintos parámetros. Basándose en la ley de Ohm se tiene que

Diferencia de potencial (V): define el campo eléctrico; la velocidad de
avance es directamente proporcional a ella.
Resistencia (R): la movilidad de las moléculas es inversamente proporcional a
ella.
Intensidad (I) : cuantifica el flujo de carga eléctrica, se relaciona
directamente con la distancia recorrida por las moléculas.
Por último, otro factor que afecta significativamente a la electroforesis es
la temperatura, esta es importante puesto que por el efecto Joule el paso de una
corriente eléctrica va a producir calor y este es directamente proporcional a la
diferencia de potencial y a la resistencia. Por lo tanto, es necesario controlar
de manera estricta la temperatura para que esta no afecte a la muestra
desnaturalizándola.
ELECTROFORESIS CAPILAR
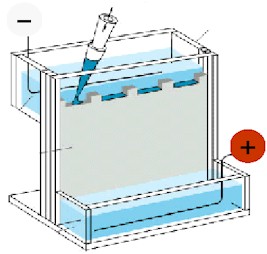
La electroforesis capilar (EC) es una técnica de separación utilizada
en distintas áreas (química, bioquímica, etc.) para separar las diferentes
moléculas presentes en una disolución de acuerdo con la relación masa/carga de
las mismas. La separación se lleva a cabo en un tubo hueco de diámetro muy
pequeño (menos de 50 µm de diámetro), de ahí que reciba el nombre de capilar.
Dentro de este capilar se encuentran la disolución que contiene los analitos o
las moléculas a separar y el tampón o medio electrolítico que es el encargado de
conducir la corriente. Específicamente, el interior se encuentra formado por
grupos silanol (Si-OH), los cuales al ser desprotonados (Si-O), elevan
considerablemente el potencial de hidrógeno (pH) y favorecen la presencia de
analitos específicos. Como se ha dicho, la separación se lleva a cabo según la
relación masa/carga de las distintas moléculas. Para que esto sea posible es
necesario aplicar una diferencia de potencial (de 100 a 500 V/cm) entre los dos
extremos del capilar que hará que las moléculas se muevan hacia un extremo u
otro del capilar (movilidad electroforética) (las moléculas catiónicas hacia el
polo negativo y las aniónicas hacia el polo positivo) y que se vayan separando
entre sí. Además, existe dentro del capilar otro fenómeno denominado flujo
electroosmotico que se da debido a que la superficie interna del capilar está
cargada. El flujo electroosmótico es el mismo dentro de todo el capilar y afecta
de igual forma a todas las moléculas arrastrándolas hacia uno de los extremos.
Así, la separación se vera afectada por el flujo electroosmótico y por la
movilidad electroforética de cada una de las moléculas.
La inducción del alto potencial eléctrico permite: 1) que la separación sea más sensible entre las diferentes moléculas (aumento de la resolución) y 2) que el tiempo de análisis sea más corto. Además, para el caso del ADN, los fragmentos a analizar se encuentran unidos a marcas fluorescentes, siendo detectadas por un láser de argón, que las excita a distintas longitudes de onda, lográndose así el análisis de múltiples fragmentos al mismo tiempo, que se van a mover hacia el polo positivo y se separarán de acuerdo a la longitud del campo eléctrico: los de menor peso molecular viajarán más rápido a través del capilar, los de mayor peso molecular lo harán más lentamente. Todas estas características convierten a la EC en un método eficiente y económico, capaz de separar cientos de componentes de forma simultánea, empleando cantidades mínimas de muestras y reactivos.
La eficacia y la velocidad de la separación se pueden mejorar mediante la optimización de diferentes factores como son la temperatura, el voltaje aplicado, el medio de separación, el disolvente en el que se encuentra disuelta la muestra, etc. Generalmente se obtienen tiempos de análisis bastante bajos si se compara con otras técnicas separativas como la cromatografía de gases o la de liquidos. Además el consumo de muestra y reactivos es muchísimo menor por lo que se la puede considerar una técnica más limpia. Es muy versátil ya que se puede emplear para separar cualquier tipo de compuesto eligiendo bien el detector.
Se puede acoplar a un detector UV, de fluorescencia, un espectrómetro de masas, etc.
En los último años, la EC ha contribuido al fortalecimiento de la ciencia y la medicina moderna. Gracias a las ventajas que provee la EC, la secuenciación del ADN ha podido automatizarse, permitiendo, hoy día, poder conocer las secuencias genómicas del hombre y otras especies con mayor velocidad y especificidad (15 mil millones de nucleótidos de secuencia en menos de un año). La información acumulada por la EC comienza a vislumbrar las causas genéticas de muchas enfermedades, fortaleciendo el diagnóstico en contraposición de las metodologías clásicas usadas para el estudio de la medicina genómica.
Las pruebas de diagnóstico genético emplean normalmente un gran número de marcadores polimórficos (variaciones específicas en la secuencia del genoma), como los "microsatélites o STR" (Short Tandem Repeat), los "minisatélites o VNTR" (Variable Number of Tandem Repeat) y los SNP (Single Nucleotide Polymorphism), además de la determinación de mutaciones asociadas a muchas enfermedades. Así, el uso de marcadores polimórficos y mutacionales para el diagnóstico requiere de una tecnología automatizada, como la EC, que contribuye, principalmente, en dos áreas del diagnóstico genético: secuenciación y análisis de fragmentos en los cuales se puede determinar la dosis génica de forma cuantitativa (como para pruebas prenatales: trisomías, monosomías, duplicaciones, deleciones e inserciones). Técnicas como la electroforesis en geles de agarosa y poliacrila-mida consumen más tiempo y recursos, son menos específicas y reproducibles, y suelen requerir mayor cantidad de muestra (actualmente se encuentran casi en desuso).

Si se han inyectado varias mezclas una junto a otra en la placa, se producirán separaciones paralelas. Cada separación mostrará distintas bandas correspondientes a cada componente de la mezcla. Si las separaciones son incompletas, se dará un solapamiento entre bandas haciendo indistinguibles dos o más componentes.
Las bandas en diferentes separaciones paralelas que están a la misma distancia del principio significa que contienen moléculas que han atravesado el gel a la misma velocidad. Existen marcadores especiales que contienen una mezcla de moléculas de tamaño conocido. Si se hace una electroforesis de un marcador con una mezcla desconocida, las bandas observadas en el marcador pueden ser comparadas con las obtenidas en la mezcla desconocida para determinar su tamaño o punto isoeléctrico. La distancia a la que se encuentra la banda del principio es (aproximadamente) inversamente proporcional al logaritmo del tamaño de la molécula.
La electroforesis proteica es la separación de proteínas mediante la aplicación de un campo
eléctrico.
Como medio de soporte se puede usar (de más antiguo a más reciente): papel, acetato de celulosa, agarosa, poliacrilamida y electroforesis capilar. La muestra cuyas proteínas se quieren separar se inserta en un medio de soporte y se aplica una diferencia de potencial durante un tiempo determinado para separar las proteínas. Cada proteína migrará más o menos en función de su carga (que también determina hacia qué polo se dirigirá la proteína, ánodo (+) o cátodo (-) y su tamaño. A mayor carga y menor tamaño, más velocidad de migración.
Isoelectroenfoque: en lugar de separar las proteínas en función de su carga a un pH dado, se separan en función de su punto isoeléctrico (pI): el pI es el pH en el que la carga neta de la proteína es nula, y depende de la composición aminoacídica de la proteína. Se crea un gradiente de pH mediante anfolitos (estabilizan el pH a lo largo del gel). Cada proteína migrará hasta alcanzar su pI, punto en el cual precipitará al acumularse (de ahí el nombre, isoelectroenfoque).
También se suelen utilizar acoplados a zimogramas si deseamos determinar el pI de una enzima o en electroforesis bidimensional como primera dimension.
Separación por tamaño: permite separar proteínas y ácidos nucleicos. En el caso de las proteínas, deben ser tratadas con SDS (sodio dodecil sulfato) para que su carga sea negativa y todas migren hacia el ánodo (no es necesario hacer eso con los ácidos nucleicos, ya que tienen carga negativa); la separación se hace en medios de soporte en el que se ha creado un tamiz molecular (matriz), que hace que las proteínas más pequeñas corran más que las más grandes.
Por lo general para la tinción de geles de poliacrilamida se utiliza tinción con plata, tinción de Coomassie o tinción fluorescente. Dependiendo del fin de nuestro análisis. La tinción con plata no es utilizada si se desean hacer análisis por espectrometría de masas (MS) pero es más sensible que la tinción con Azul de Coomassie, por otro lado, la tinción fluorescente es muy sensible y compatible con MS pero los costos de tinción son elevados.
Una tinción sensible y barata es la denominada "Blue Silver" o Coomassie G250.[1] Además, esta tinción es compatible con MS. Esta compuesta por azul de coomassie diluido en una solución coloidal y se utiliza agua destilada para destiñir el gel de poliacrilamida.
Este tipo de análisis electroforético tiene aplicaciones en investigación y en clínica, tanto humana como animal. Además es una técnica muy empleada para el análisis de proteínas alimentarias y últimamente se empleando para realizar genotipado y detección de OMG (organismos modificados genéticamente)
La inducción del alto potencial eléctrico permite: 1) que la separación sea más sensible entre las diferentes moléculas (aumento de la resolución) y 2) que el tiempo de análisis sea más corto. Además, para el caso del ADN, los fragmentos a analizar se encuentran unidos a marcas fluorescentes, siendo detectadas por un láser de argón, que las excita a distintas longitudes de onda, lográndose así el análisis de múltiples fragmentos al mismo tiempo, que se van a mover hacia el polo positivo y se separarán de acuerdo a la longitud del campo eléctrico: los de menor peso molecular viajarán más rápido a través del capilar, los de mayor peso molecular lo harán más lentamente. Todas estas características convierten a la EC en un método eficiente y económico, capaz de separar cientos de componentes de forma simultánea, empleando cantidades mínimas de muestras y reactivos.
La eficacia y la velocidad de la separación se pueden mejorar mediante la optimización de diferentes factores como son la temperatura, el voltaje aplicado, el medio de separación, el disolvente en el que se encuentra disuelta la muestra, etc. Generalmente se obtienen tiempos de análisis bastante bajos si se compara con otras técnicas separativas como la cromatografía de gases o la de liquidos. Además el consumo de muestra y reactivos es muchísimo menor por lo que se la puede considerar una técnica más limpia. Es muy versátil ya que se puede emplear para separar cualquier tipo de compuesto eligiendo bien el detector.
Se puede acoplar a un detector UV, de fluorescencia, un espectrómetro de masas, etc.
En los último años, la EC ha contribuido al fortalecimiento de la ciencia y la medicina moderna. Gracias a las ventajas que provee la EC, la secuenciación del ADN ha podido automatizarse, permitiendo, hoy día, poder conocer las secuencias genómicas del hombre y otras especies con mayor velocidad y especificidad (15 mil millones de nucleótidos de secuencia en menos de un año). La información acumulada por la EC comienza a vislumbrar las causas genéticas de muchas enfermedades, fortaleciendo el diagnóstico en contraposición de las metodologías clásicas usadas para el estudio de la medicina genómica.
Las pruebas de diagnóstico genético emplean normalmente un gran número de marcadores polimórficos (variaciones específicas en la secuencia del genoma), como los "microsatélites o STR" (Short Tandem Repeat), los "minisatélites o VNTR" (Variable Number of Tandem Repeat) y los SNP (Single Nucleotide Polymorphism), además de la determinación de mutaciones asociadas a muchas enfermedades. Así, el uso de marcadores polimórficos y mutacionales para el diagnóstico requiere de una tecnología automatizada, como la EC, que contribuye, principalmente, en dos áreas del diagnóstico genético: secuenciación y análisis de fragmentos en los cuales se puede determinar la dosis génica de forma cuantitativa (como para pruebas prenatales: trisomías, monosomías, duplicaciones, deleciones e inserciones). Técnicas como la electroforesis en geles de agarosa y poliacrila-mida consumen más tiempo y recursos, son menos específicas y reproducibles, y suelen requerir mayor cantidad de muestra (actualmente se encuentran casi en desuso).
ELECTROFORESIS EN GEL
La electroforesis en gel es un grupo de técnicas empleadas por los científicos para separar moléculas basándose en propiedades como el tamaño, la forma o el punto isoeléctrico. La electroforesis en gel se utiliza generalmente con propósitos analíticos, pero puede ser una técnica preparativa para purificar moléculas parcialmente antes de aplicar espectrometría de masas, PCR, clonación o secuenciación de ADN
Aplicaciones
Proteínas
Las proteínas no tienen una estructura predecible como los ácidos nucleicos, y por tanto sus velocidades de migración no son similares entre ellas. Incluso puede que no migren ni al aplicar una fuerza electromotriz (al encontrarse en su punto isoeléctrico). En estos casos, las proteínas se desnaturalizan mediante la adición de un detergente como el dodecilsulfato sódico/dodecilfosfato sódico (SDS/SDP) y un agente reductor como el 2-mercaptoetanol. Los detergentes otorgan una carga neta negativa a la proteína que les permite migrar a través del gel de poliacrilamida en relación directa a su masa, ya que la cantidad de cargas negativas que se unen a la proteína depende del tamaño de ésta, existiendo una relación carga/masa similar. Por otro lado, el agente reductor rompe los enlaces disulfuros, separando a la proteína en sus sub-unidades. Además, la desnaturalización hace que pierdan su estructura terciaria y cuaternaria por tanto su velocidad de migración es proporcional al tamaño y no a su estructura terciaria ni a su interacción con otras macromoleculas. Así, los más grandes se desplazan más lentamente.Revelado y visualización
Cuando se ha completado la electroforesis, las moléculas más pequeñas han llegado al ánodo. Entonces se pueden 'revelar' mediante la adición de un colorante específico para hacerlas visibles. Se emplean compuestos como el bromuro de etidio, para los ácidos nucleicos, o tinción de plata, azul de coomassie o tinción fluorescente, para las proteínas. Asimismo se emplean otros métodos para visualizar la separación de la mezcla en el gel. Si el reactivo es fluorescente bajo la luz UV, se puede simplemente hacer una fotografía de la placa bajo dicha luz. También, si las moléculas contienen átomos radiactivos se puede efectuar una autorradiografía.Si se han inyectado varias mezclas una junto a otra en la placa, se producirán separaciones paralelas. Cada separación mostrará distintas bandas correspondientes a cada componente de la mezcla. Si las separaciones son incompletas, se dará un solapamiento entre bandas haciendo indistinguibles dos o más componentes.
Las bandas en diferentes separaciones paralelas que están a la misma distancia del principio significa que contienen moléculas que han atravesado el gel a la misma velocidad. Existen marcadores especiales que contienen una mezcla de moléculas de tamaño conocido. Si se hace una electroforesis de un marcador con una mezcla desconocida, las bandas observadas en el marcador pueden ser comparadas con las obtenidas en la mezcla desconocida para determinar su tamaño o punto isoeléctrico. La distancia a la que se encuentra la banda del principio es (aproximadamente) inversamente proporcional al logaritmo del tamaño de la molécula.
Tipos
La electroforesis en gel se utiliza en biología molecular, genética y bioquímica:- La electroforesis en gel de muestras grandes de ADN y ARN se efectúa en geles de agarosa.
- La electroforesis de proteínas se lleva a cabo en geles de poliacrilamida-SDS (SDS-PAGE), isoelectroenfoque, geles nativos o electroforesis bidimensional.
- Electroforesis capilar.
- Electroforesis de ADN.
- Zimografía o zimogramas
- Extracción en gel.
ELECTROFORESIS PROTEICA
La electroforesis proteica es la separación de proteínas mediante la aplicación de un campo
eléctrico.
Existen diferentes tipos en función del tipo de separación empleado:
electroforesis de zona (separación en función de la carga), isoelectroenfoque y
separación por tamaño en tamiz molecular (también aplicable a ácidos
nucleicos).
Técnicas
Electroforesis de zona: las proteínas son moléculas anfóteras: su carga neta depende del pH del medio. Normalmente, la separación electroforética de proteínas se hace a pH alcalino, en el que la mayoría de las proteínas presentan una carga global negativa. También se puede trabajar a pH ácidos, pero no demasiado bajos, ya que las proteínas precipitan en medio ácido (básicamente se usa en la detección de variantes de la hemoglobina).Como medio de soporte se puede usar (de más antiguo a más reciente): papel, acetato de celulosa, agarosa, poliacrilamida y electroforesis capilar. La muestra cuyas proteínas se quieren separar se inserta en un medio de soporte y se aplica una diferencia de potencial durante un tiempo determinado para separar las proteínas. Cada proteína migrará más o menos en función de su carga (que también determina hacia qué polo se dirigirá la proteína, ánodo (+) o cátodo (-) y su tamaño. A mayor carga y menor tamaño, más velocidad de migración.
Isoelectroenfoque: en lugar de separar las proteínas en función de su carga a un pH dado, se separan en función de su punto isoeléctrico (pI): el pI es el pH en el que la carga neta de la proteína es nula, y depende de la composición aminoacídica de la proteína. Se crea un gradiente de pH mediante anfolitos (estabilizan el pH a lo largo del gel). Cada proteína migrará hasta alcanzar su pI, punto en el cual precipitará al acumularse (de ahí el nombre, isoelectroenfoque).
También se suelen utilizar acoplados a zimogramas si deseamos determinar el pI de una enzima o en electroforesis bidimensional como primera dimension.
Separación por tamaño: permite separar proteínas y ácidos nucleicos. En el caso de las proteínas, deben ser tratadas con SDS (sodio dodecil sulfato) para que su carga sea negativa y todas migren hacia el ánodo (no es necesario hacer eso con los ácidos nucleicos, ya que tienen carga negativa); la separación se hace en medios de soporte en el que se ha creado un tamiz molecular (matriz), que hace que las proteínas más pequeñas corran más que las más grandes.
Visualización
Una vez separadas las proteínas, deben fijarse y teñirse para poder ser visualizadas. Hay diferentes protocolos en función de lo que se quiere estudiar: fijación por calor o química y tinción en caso de estudios no específicos (proteinograma y electroforesis Hb, por ejemplo) o fijación mediante anticuerpos previa a la tinción en caso de estudiar proteínas específicas (inmunofijación).Por lo general para la tinción de geles de poliacrilamida se utiliza tinción con plata, tinción de Coomassie o tinción fluorescente. Dependiendo del fin de nuestro análisis. La tinción con plata no es utilizada si se desean hacer análisis por espectrometría de masas (MS) pero es más sensible que la tinción con Azul de Coomassie, por otro lado, la tinción fluorescente es muy sensible y compatible con MS pero los costos de tinción son elevados.
Una tinción sensible y barata es la denominada "Blue Silver" o Coomassie G250.[1] Además, esta tinción es compatible con MS. Esta compuesta por azul de coomassie diluido en una solución coloidal y se utiliza agua destilada para destiñir el gel de poliacrilamida.
Aplicaciones
Pueden analizarse las proteínas contenidas en diferentes líquidos biológicos: sangre, plasma (el líquido sanguíneo sin células), suero (plasma sin fibrinógeno), orina, LCR, líquido sinovial, saliva, lágrimas. Así como alimentos, especialmente lácteos y cereales.Este tipo de análisis electroforético tiene aplicaciones en investigación y en clínica, tanto humana como animal. Además es una técnica muy empleada para el análisis de proteínas alimentarias y últimamente se empleando para realizar genotipado y detección de OMG (organismos modificados genéticamente)
- Bandow J, Baker JD, Berth M, Painter C, et al.: Improved image analysis workflow for 2-D gels enables large-scale 2-D gel-based proteomics studies - COPD biomarker discovery study. Proteomics 2008 [1]
- Berth M, Moser FM, Kolbe M, et al: The state of the art in the analysis of two-dimensional gel eletcrophoresis images. Appl Microbiol Biotechnol. 2007;76(6):1223–43
- Candiano, G., Bruschi, M., Musante, L., Santucci, L., Ghiggeri, G.M., Carnemolla, B., Orecchia, P., Zardi, L. and Righetti, P.G. (2004). «Blue silver: A very sensitive colloidal Coomassie G-250 staining for proteome analysis.». Electrophoresis, 25 (9): pp. 1327 - 1333
No hay comentarios:
Publicar un comentario