
La presión de vapor es la presión de la fase gaseosa o vapor de un sólido o un líquido sobre la fase líquida, para una temperatura determinada, en la que la fase líquida y el vapor se encuentra en equilibrio dinámico; su valor es independiente de las cantidades de líquido y vapor presentes mientras existan ambas. Este fenómeno también lo presentan los sólidos; cuando un sólido pasa al estado gaseoso sin pasar por el estado líquido (proceso denominado sublimación o el proceso inverso llamado deposicitación o sublimación inversa) también hablamos de presión de vapor. En la situación de equilibrio, las fases reciben la denominación de líquido saturado y vapor saturado. Esta propiedad posee una relación directamente proporcional con las fuerzas de atracción intermoleculares, debido a que cuanto mayor sea el módulo de las mismas, mayor deberá ser la cantidad de energía entregada (ya sea en forma de calor u otra manifestación) para vencerlas y producir el cambio de estado.
Imaginemos una burbuja de cristal en la que se ha realizado el vacío y que se mantiene a una temperatura constante; si introducimos una cierta cantidad de líquido en su interior éste se evaporará rápidamente al principio hasta que se alcance el equilibrio entre ambas fases.
Inicialmente sólo se produce la evaporación ya que no hay vapor; sin embargo a medida que la cantidad de vapor aumenta y por tanto la presión en el interior de la ampolla, se va incrementando también la velocidad de condensación, hasta que transcurrido un cierto tiempo ambas velocidades se igualan. Llegados a este punto se habrá alcanzado la presión máxima posible en la ampolla (presión de vapor o de saturación) que no podrá superarse salvo que se incremente la temperatura.

El equilibrio dinámico se alcanzará más rápidamente cuanto mayor sea la superficie de contacto entre el líquido y el vapor, pues así se favorece la evaporación del líquido; del mismo modo que un charco de agua extenso pero de poca profundidad se seca más rápido que uno más pequeño pero de mayor profundidad que contenga igual cantidad de agua. Sin embargo, el equilibrio se alcanza en ambos casos para igual presión.
El factor más importante que determina el valor de la presión de saturación es la propia naturaleza del líquido, encontrándose que en general entre líquidos de naturaleza similar, la presión de vapor a una temperatura dada es tanto menor cuanto mayor es el peso molecular del líquido.
Por ejemplo, el aire al nivel del mar saturado con vapor de agua a 20ºC, tiene una presión parcial de 23 mbar de agua y alrededor de 780 mbar de nitrógeno, 210 mbar de oxígeno y 9 mbar de argon.
Medición y unidades
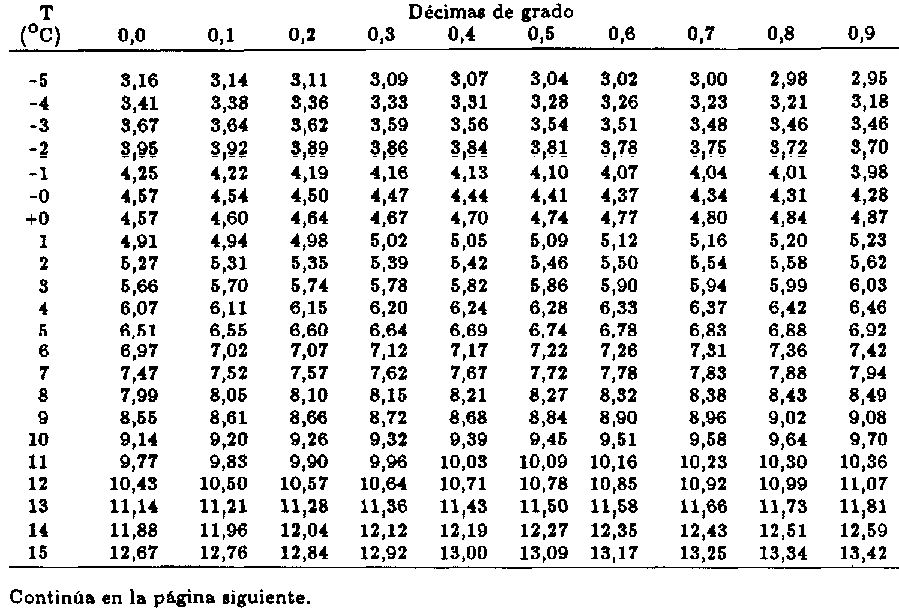
La medición experimental de la presión de vapor es un procedimiento simple para presiones similares que estén entre 1 y 200 kPa. Resultados más exactos son obtenidos cerca del punto de ebullición de cada sustancia en particular y con índice de error más significativo en mediciones menores a 1 kPa. Con frecuencia, algunos procedimientos consisten en purificar las sustancias que son analizadas, aislándolas la sustancia deseada en un contenedor, evitando cualquier gas indeseado y midiendo la presión de equilibrio de la fase gaseosa de la sustancia en el sistema cerrado a distintas temperaturas. El uso de herramientas como un isoteniscopio genera una mayor exactitud en el proceso.

La regla de fases establece que la presión del vapor de un líquido puro es función única de la temperatura de saturación. Vemos pues que la presión de vapor en la mayoría de los casos se puede expresar como
Pvp
= f (t)
La cual podría estar
relacionada con cualquier otra propiedad intensiva de un líquido saturado ( o
vapor), pero es mucho mejor relacionarla directamente con la temperatura de
saturaciónLa presión de vapor de un liquido se relaciona con la temperatura por medio de la ecuación de Claussius Clapeyron, sin embargo existen muchas ecuaciones que estudian esta propiedad de los fluidos, pero de todas maneras estas ecuaciones pueden referirse a la ecuación de Clapeyron:
Ln
P2/P1 = (DH/R) vaporización (1/T1-1/T2)
Esta ecuación mediante pasos
matemáticos, puede convertirse en:
Ln
Pvp = A+B/T
La gráfica del logaritmo de la presión del vapor y el
reciproco de la temperatura absoluta es una recta. La ecuación anterior no es
una mala aproximación pero en general esta curva realmente tiene unas
curvaturas pequeñas que muestran así que esta aproximación tampoco es la mejor.
Estas curvas las observamos exagerando un poco el dibujo, de la siguiente
manera- En intervalos de baja presión:
10 a 1500 mmHg se estima por varios métodos unos de los cuales son:
El método de estimación de Frost-Kalkwarf-Thodors, es
el mejor para compuestos orgánicos, el cual se hace por medio de
Cálculos de tipo iterativo, y arroja un máximo porcentaje de error medio
de 5.1%
El método de Riedel-Plank-Miller es el mejor para
compuestos inorgánicos y además es fácil de usar, este arroja un máximo
porcentaje de error medio de 5.2%
- En intervalos de alta presión:
1500 mmHg hasta la presión critica también existen varios métodos de los
cuales mencionare algunos:
El método de estimación reducida de Kirchhoff, el cual
no es muy exacto pero es muy fácil de usar, este arroja un máximo porcentaje de
error medio de 3.2%
El método de estimación de Frost-Kalkwarf-Thodors,
para intervalos de alta presión también
requiere de cálculos iterativos, sin embargo es muy bueno y arroja un máximo porcentaje de error medio de
1.5%
Estos métodos anteriores son métodos trabajados con
ecuaciones reducidas para los cuales era
necesario conocer tc, pc, tb.. pero existen
muchísimos método diferentes tanto con
ecuaciones reducidas como con ecuaciones semirreducidas y sin reducir.
¿Cuál es algún tipo de uso de la presión de vapor?
Para mirar
un ejemplo de presión de vapor aplicada a tuberías es bueno analizar un poco
las plantas productoras de petroquímicos y refinerías, ya que estas requieren
de muchos servicios como: vapor de agua (enfriamiento, servicio, proceso), aire
de instrumentos, energía eléctrica; para ello estas plantas necesitan grandes
sistemas de transformación de energía, y redes de distribución de varios
kilómetros, en las cuales se incurre en perdidas de energía. Para lo que es
necesario usar expresiones matemáticas para calcular dichas perdidas y llevar a
cabo estudios sobre la recuperación de la inversión y la rentabilidad de acciones de ahorro de
energía.
Se debe realizar un pequeño análisis de los sistemas
de generación y distribución de vapor, principalmente de aquellos que por ser
de gran tamaño son muy dinámicos cambiando sus condiciones de operación; flujo,
temperatura y presión varias veces al día. Los cambios pueden ser ocasionados
por modificación en las condiciones de operación de las plantas de proceso de
mantenimiento predictivo o correctivo de los equipos generadores y consumidores
de vapor y energía eléctrica, o por cambio de las condiciones atmosféricas.
Estos cambios nos proporcionan áreas de oportunidad de ahorro si se mantiene un
análisis constante del sistema de generación y distribución de vapor.
En la generación del vapor vemos como las plantas que
lo generan, están formadas por dos o tres niveles de presión, los cuales son
distribuidos según su uso o según la magnitud de la presión del vapor, de esta
forma: para los bloques de generación eléctrica, turbinas para accionar bombas
y compresores de plantas de procesos se usa el vapor de mayor presión; para
turbogeneradores eléctricos y grandes turbocompresores, se usa por lo general
extracciones de vapor media; las turbinas de menor capacidad normalmente
descargan a la red de baja presión.

El control de la presión y la temperatura en las redes
de distribución de vapor es sumamente importante, ya que excesos de estas
presiones pueden causar un desgaste mas acelerado de la tubería y aparte de
esto se pueden generar muchas perdidas de energía, lo cual no es conveniente
para un proceso en el cual se esta tratando de aprovechar la energía al máximo.
Para controlar estos excesos o simplemente variantes
de las presiones y temperaturas adecuadas se tienen controles de los
generadores de vapor los cuales
mantienen estos factores en los valores ajustados, esta regulación también se lleva a cabo durante todo el proceso ya
que en las redes de media y baja presión, también se cuenta con reguladores de
presión y temperatura en turbinas y otros aparatos que intervienen en el este.
Ya con estos reguladores en el procesos, se puede
decir que cuando las condiciones de presión y temperatura del vapor que llegan
a los equipos varían. La demanda de vapor se ajustará dependiendo de la
entalpía y otras características del vapor y del salto entalpico disponible, en
el caso de las turbinas.

Para mantener el control en los sistemas de
distribución de vapor, es necesario llevar una buena administración y una
constante revisión de toda la red, a su ves mediante los dato recolectado
durante las revisiones periódicas es necesario estar calculando las perdidas de
energía ya que estas afecten directamente la eficiencia del proceso, por ultimo
es necesario determinar los puntos de ajuste adecuados para la red.
También es necesario analizar la posibilidad de operar
la red a la menor presión posible para lo cual seria necesario consultar a los
fabricantes de las turbinas y determinar si la tubería podría transportar los
volúmenes necesarios.
La hoja de un patín para hielo está apoyada sobre el filo
de una cuchilla.
a) Si
el ancho del filo de la cuchilla es de 2.54 x10-3 cm y el largo del patín en contacto con el hielo es de 7.62
cm, calcular la presión ejercida sobre el hielo por un hombre que pesa 68
kg.
b)
¿Cuál es el punto de fusión del hielo bajo esta presión?
(DHfus = 1.4363 kcal/mol, Tf = 273 K, rs = 0.92 g/cm3 y rl = 1.00
g/cm3).
Solución:
Se trata de un equilibrio sólido - líquido, aplicar
la ecuación de Clapeyron:

Area de la cuchilla: (2.54
x10-3) (7.62) = 0.0193 cm2.
Presión que ejerce el patinador:
P = 3409.8 atm.
Como se
proporcionan las densidades del sólido y del líquido, se pueden calcular los
volúmenes molares del agua (tomando una masa igual a su peso molecular, PM = 18
g/mol).
P1 = 1 atm; T1 = 0 ºC = 273
K.
P2 = 3409.8 atm; T2 = ?.
T2 = 249.5 K = -23.49
ºC
1. La presión de vapor del bromobenceno es 1
mmHg a 2.9 ºC y de 20 mmHg a 53.8
ºC. Calcular el punto de ebullición normal. ¿Cuáles serán las posibles
razones para las diferencias con el valor experimental de 156.2 ºC?
Resp. T = 421 K = 148 ºC.
2. El hielo II y III están en equilibrio a
–25 ºC y 3260 atm y a -31 ºC y 2540 atm. Sus densidades respectivas son 1.21 y
1.1g/cm3. Calcular el DH para la
transición de 1 mol de hielo II a hielo III.
Resp. DHcf = 1069.1
cal/mol.
3. El azufre rómbico (Sr) y el monoclínico
(Sm) están en equilibrio a 115 ºC y 1atm, y a 120 ºC y 100 atm. El DH de transición de Sr a Sm es de 70 Kcal. ¿Cuál forma alotrópica
tiene mayor densidad?
Resp. DV = 371 cm3; implica que: Vm
> Vc, por lo tanto: dr
> dm.
4. El iodo hierve a 183 ºC a 760 mmHg. La presión de vapor
a 116 ºC es de 100 mmHg. El calor de fusión es de 3740 cal/mol y la presión de
vapor del sólido es de 1 mmHg a 38.7 ºC. Calcular la temperatura y la presión
del punto triple.
Resp. T3 = 384.8 K; P3 = 84.77
mmHg.
5. El agua y el cloroformo tienen una temperatura de
ebullición normal de 100 y 60
ºC, respectivamente y un DH de evaporación de 12 y 7
kcal/mol. Calcular la temperatura a la cual los dos tienen la misma presión de
vapor.
Resp. T = 447.23 K.
PRESION OSMÓTICA

Esquema de una membrana semipermeable. Las moléculas
grandes de la sangre no pueden
atravesar la membrana, mientras que las pequeñas de solvente sí.
La presión osmótica puede definirse como la presión que se debe aplicar a una solución para detener el flujo neto de disolvente a través de una membrana semipermeable.[1] La presión osmótica es una de las cuatro propiedades coligativas de las soluciones (dependen del número de partículas en disolución, sin importar su naturaleza). Se trata de una de las características principales a tener en cuenta en las relaciones de los líquidos que constituyen el medio interno de los seres vivos, ya que la membrana plasmática regula la entrada y salida de soluto al medio extracelular que la rodea, ejerciendo de barrera de control.
Cuando dos soluciones se ponen en contacto a través de una membrana semipermeable (membrana que deja pasar las moléculas de disolvente pero no las de los solutos), las moléculas de disolvente se difunden, pasando habitualmente desde la solución con menor concentración de solutos a la de mayor concentración. Este fenómeno recibe el nombre de ósmosis, palabra que deriva del griego osmos, que significa "impulso".[2] Al suceder la ósmosis, se crea una diferencia de presión en ambos lados de la membrana semipermeable: la presión osmótica.

Esquema del modo de acción de la presión osmótica. En azul
se representan las moléculas de disolvente y en rojo las de soluto. La disolución más concentrada se denomina hipertónica y la diluida hipotónica. Como consecuencia
de la diferencia inicial de concentraciones se produce una presión osmótica,
apareciendo una diferencia de altura h, hasta que las concentraciones se
igualan.
Se considera que una disolución está en equilibrio cuando no existe intercambio neto de
soluto entre las diferentes partes de la misma. Si la disolución se encuentra
rodeada por una membrana, el equilibrio se alcanza cuando la presión exterior
(generalmente la presión atmosférica) se iguala a
la presión que el disolvente ejerce sobre la membrana. Ésta última es la presión
osmótica, que se representa habitualmente mediante la letra griega Π.
Cuando se tiene una membrana semipermeable separando dos soluciones de distinta concentración (llamada hipertónica a la de mayor concentración e hipotónica la de menor), las moléculas de disolvente (agua por lo general) la atraviesan, pasando de la disolución menos concentrada a la más concentrada, diluyéndose ésta última cada vez más, hasta que las concentraciones se igualen. Si el volumen era inicialmente idéntico en las dos soluciones, ocurre que en la solución hipertónica el volumen aumenta, hasta que la presión hidrostática (que aumenta debido al incremento de altura h) iguale las presiones a ambos lados de la membrana. Esta presión hidrostática que detiene el flujo neto de disolvente es equivalente a la presión osmótica, y es el fundamento del osmómetro utilizado para su medición.
Las primeras investigaciones sobre la presión osmótica fueron realizadas en
1748 por el abad francés Jean Antoine
Nollet, cuando era profesor de física en la Universidad
de Navarra, quien descubrió la existencia de las membranas
semipermeables. Nollet obtuvo una membrana a partir de una vejiga de cerdo, colocó
alcohol a un lado y agua al otro, y observó que el agua fluía a través de la
vejiga para mezclarse con el alcohol, pero el alcohol no lo hacía.[3]
No obstante, el descubrimiento de la ósmosis en membranas semipermeables se le atribuye a Henri Dutrochet, considerado uno de los grandes fisiólogos del siglo XIX, en el año 1828.[4] Dutrochet descubrió este fenómeno al observar que la difusión de disolvente a través de una membrana semipermeable ocurría siempre desde la disolución de menor concentración de un soluto, el cual no podía atravesarla, hasta la disolución de mayor concentración; además, el disolvente que fluye es capaz de ejercer una presión sobre la membrana, la presión osmótica. Dutrochet construyó el primer dispositivo experimental para observar la presencia de la presión osmótica, denominado osmómetro. Al descubrir la ósmosis declaró:
El inglés Thomas Graham descubrió, en 1854, que las sustancias coloidales no atravesaban ciertas membranas. Sin embargo, los primeros estudios cuantitativos datan de 1877 y se deben al alemán Wilhelm Pfeffer,[5] profesor de botánica en Tubingen y Leipzig, quien preparó una membrana semipermeable artificial precipitando hexacianoferrato (II) de cobre (II), Cu2[Fe(CN)6], sobre las paredes de un vaso poroso.[6] Pfeffer, a partir de la medición de presiones osmóticas en multitud de disoluciones de solutos no volátiles, llegó a las siguientes conclusiones:
El holandés Jacobus Henricus van 't Hoff realizó un estudio sistemático de las propiedades coligativas de las disoluciones, que publicó en 1885.[8] En este artículo van 't Hoff formula una expresión, para disoluciones diluidas, que relaciona la presión osmótica con la concentración del soluto, la cual es similar a la ecuación de los gases ideales y proporciona la primera teoría para explicar la presión osmótica:

donde:

La presión osmótica puede definirse como la presión que se debe aplicar a una solución para detener el flujo neto de disolvente a través de una membrana semipermeable.[1] La presión osmótica es una de las cuatro propiedades coligativas de las soluciones (dependen del número de partículas en disolución, sin importar su naturaleza). Se trata de una de las características principales a tener en cuenta en las relaciones de los líquidos que constituyen el medio interno de los seres vivos, ya que la membrana plasmática regula la entrada y salida de soluto al medio extracelular que la rodea, ejerciendo de barrera de control.
Cuando dos soluciones se ponen en contacto a través de una membrana semipermeable (membrana que deja pasar las moléculas de disolvente pero no las de los solutos), las moléculas de disolvente se difunden, pasando habitualmente desde la solución con menor concentración de solutos a la de mayor concentración. Este fenómeno recibe el nombre de ósmosis, palabra que deriva del griego osmos, que significa "impulso".[2] Al suceder la ósmosis, se crea una diferencia de presión en ambos lados de la membrana semipermeable: la presión osmótica.
Presión osmótica de equilibrio

Cuando se tiene una membrana semipermeable separando dos soluciones de distinta concentración (llamada hipertónica a la de mayor concentración e hipotónica la de menor), las moléculas de disolvente (agua por lo general) la atraviesan, pasando de la disolución menos concentrada a la más concentrada, diluyéndose ésta última cada vez más, hasta que las concentraciones se igualen. Si el volumen era inicialmente idéntico en las dos soluciones, ocurre que en la solución hipertónica el volumen aumenta, hasta que la presión hidrostática (que aumenta debido al incremento de altura h) iguale las presiones a ambos lados de la membrana. Esta presión hidrostática que detiene el flujo neto de disolvente es equivalente a la presión osmótica, y es el fundamento del osmómetro utilizado para su medición.
Historia

No obstante, el descubrimiento de la ósmosis en membranas semipermeables se le atribuye a Henri Dutrochet, considerado uno de los grandes fisiólogos del siglo XIX, en el año 1828.[4] Dutrochet descubrió este fenómeno al observar que la difusión de disolvente a través de una membrana semipermeable ocurría siempre desde la disolución de menor concentración de un soluto, el cual no podía atravesarla, hasta la disolución de mayor concentración; además, el disolvente que fluye es capaz de ejercer una presión sobre la membrana, la presión osmótica. Dutrochet construyó el primer dispositivo experimental para observar la presencia de la presión osmótica, denominado osmómetro. Al descubrir la ósmosis declaró:
Este descubrimiento que he hecho
pertenece a una clase nueva de fenómenos físicos que sin duda alguna intervienen
fuertemente en los procesos vitales.[3]
De este modo, Dutrochet intuyó la importancia del fenómeno en las células, las cuales absorberían o
retendrían determinadas sustancias.El inglés Thomas Graham descubrió, en 1854, que las sustancias coloidales no atravesaban ciertas membranas. Sin embargo, los primeros estudios cuantitativos datan de 1877 y se deben al alemán Wilhelm Pfeffer,[5] profesor de botánica en Tubingen y Leipzig, quien preparó una membrana semipermeable artificial precipitando hexacianoferrato (II) de cobre (II), Cu2[Fe(CN)6], sobre las paredes de un vaso poroso.[6] Pfeffer, a partir de la medición de presiones osmóticas en multitud de disoluciones de solutos no volátiles, llegó a las siguientes conclusiones:
- 1. A presión constante, la presión osmótica es directamente proporcional a la concentración de soluto.
- 2. La presión osmótica de una concentración determinada es directamente proporcional a la temperatura.
- 3. A una temperatura determinada, dos disoluciones con el mismo número de moles tienen la misma presión osmótica.
El holandés Jacobus Henricus van 't Hoff realizó un estudio sistemático de las propiedades coligativas de las disoluciones, que publicó en 1885.[8] En este artículo van 't Hoff formula una expresión, para disoluciones diluidas, que relaciona la presión osmótica con la concentración del soluto, la cual es similar a la ecuación de los gases ideales y proporciona la primera teoría para explicar la presión osmótica:
donde:
- Π, es la presión osmótica, en atmósferas (atm).
- R, es la constante universal de los gases ideales (aunque a pesar de su nombre no sólo se aplica a gases, como es el caso). Su valor es de 0,082 atm·L·K-1·mol-1.
- T, es la temperatura absoluta, en kelvin (K).
- c, es la concentración molar o molaridad de la disolución, medida en mol·L-1.
Teoría de van 't Hoff
Se han propuesto diversas teorías para explicar la causa de la ósmosis. La primera teoría fue la del bombardeo de van 't Hoff, que está basada en la analogía entre la ecuación de la presión osmótica y la ley de los gases ideales. Van 't Hoff describió la presión osmótica como el resultado de las colisiones de las moléculas de soluto contra la membrana semipermeable, y supuso que las moléculas de disolvente no contribuían de ninguna manera. Con este modelo, la presión osmótica de una disolución es la misma presión que un gas ideal ejercería si ocupase el mismo volumen de la disolución.
Las partículas de un gas se mueven en todas direcciones y chocan entre ellas mismas y con las paredes del recipiente. Los choques contra las paredes representan la presión del gas. En esta animación el disolvente se representa con bolas azules y el soluto con bolas rojas.
Otras teorías
La suposición en la teoría de van 't Hoff de que las moléculas del disolvente no ejercen ningún efecto sobre la presión osmótica constituye un interrogante, ya que estas partículas se encuentran golpeando continuamente la membrana semipermeable. De este problema surge otra teoría, que considera el bombardeo de las moléculas de disolvente; según esta teoría las moléculas de disolvente bombardean la membrana semipermeable de manera desigual y provocan la ósmosis y la presión osmótica, aunque la diferencia de presiones aumente con la cantidad de soluto.
Una tercera teoría explica la ósmosis en base al descenso de la presión de vapor, el cual da lugar a que el disolvente se difunda a través de la membrana hasta que se igualan las dos presiones. Si se aplica una presión igual a la presión osmótica de la disolución se saturará la destilación, y se necesitaría una presión mayor para invertir la dirección.[6] [9] El flujo de disolvente a través de una membrana semipermeable es análogo al flujo de disolvente en la fase vapor que se produce si se dejan, dentro de un recipiente cerrado, muestras del disolvente y de una disolución. Como la presión de vapor del disolvente es mayor, se produce una transferencia neta de disolvente hacia la disolución. El flujo de disolvente continúa hasta que todo el disolvente ha pasado a la disolución.[10]
Magnitud de la presión osmótica
La presión osmótica, como su nombre indica, es una presión, y por tanto tiene las mismas unidades que el resto de presiones, es decir, Pascales (Pa) en el Sistema Internacional, aunque tradicionalmente también se utilizan las atmósferas (atm).
La molaridad mide la cantidad de masa del soluto por volumen de disolución. La molaridad y la presión osmótica son dos magnitudes relacionadas proporcionalmente; el aumento o disminución de una de ellas produce el mismo efecto en la otra, aunque en distinta proporción. Del mismo modo, la temperatura (medida en kelvin, K) también posee la misma relación con la presión osmótica.
A continuación se muestra una tabla con los valores de la presión osmótica correspondientes a diferentes concentraciones de sacarosa a una temperatura constante de 293 K (20 °C). La concentración se expresa en molalidad y no en molaridad, según la ecuación de Morse, pero las diferencias son mínimas.
Presión osmótica experimental de disoluciones de sacarosa a 20 °C[11] Molalidad (g sacarosa/kg agua) 0,1 1,0 2,0 3,0 4,0 6,0 Presión osmótica (atm) 2,47 27,2 58,4 95,2 139,0 232,3
Comparación entre presión osmótica y presión hidrostática
Al tratarse ambas de presiones, se puede comparar el valor de la presión osmótica con el de la presión hidrostática en determinadas situaciones. La presión hidrostática viene dada por:

Con esta fórmula, se puede comparar a qué profundidad de agua correspondería cualquier valor de la presión osmótica de la tabla anterior. Por ejemplo, a molalidad 1 gsac/kgagua corresponde una presión de 27,2 atm. De este modo, despejando h:

 2 750 000 Pa), obtenemos finalmente la altura:
2 750 000 Pa), obtenemos finalmente la altura:

La presión osmótica como proceso termodinámico irreversible
Desde el punto de vista de la física, en un sistema binario no reaccionante, en que los componentes no acarrean carga eléctrica y existe una temperatura uniforme e igual para dos reservoirs, se tiene que la producción de entropía es:[12]
Relación entre presión osmótica y entropía
Desde el punto de vista de la termodinámica, se puede relacionar la presión osmótica con la entropía para explicar el sentido de flujo del disolvente. El paso de disolvente hacia la disolución representa un aumento de entropía del sistema. Las moléculas de soluto aumentan su desorden al diluir la disolución mezclándose con el disolvente que atraviesa la membrana, ya que las moléculas de soluto tienen más espacio para moverse, lo que representa un mayor número de posiciones disponibles (mayor desorden), y, por tanto, una mayor entropía. También las moléculas de disolvente tienen mayor desorden en una disolución que en estado puro. Si el disolvente pasase desde la disolución al disolvente puro el orden aumentaría, lo cual disminuiría la entropía y no se cumpliría el segundo principio de la termodinámica. Todo esto demostrable matemáticamente.[10]
Relaciones con otras magnitudes físicas y químicas
Disoluciones diluidas de no electrolitos
En el caso de disoluciones diluidas de no electrolitos se aplica directamente la ecuación de van 't Hoff. Si la presión osmótica se produce entre dos disoluciones, en lugar de entre una disolución y el disolvente puro, en la ecuación figura la diferencia de concentraciones, ΔC:


[mostrar]Deducción termodinámica de las ecuaciones de Morse y van 't Hoff[14] Disoluciones diluidas de electrolitos
Jacobus Henricus van 't Hoff descubrió que las disoluciones de electrolitos no cumplían la ecuación de la presión osmótica para disoluciones de no electrolitos, e introdujo el llamado factor de van 't Hoff, un factor determinado empíricamente simbolizado por la letra ,
y definido como el cociente entre el valor experimental de la presión osmótica
media y el valor teórico que se deduce con la ecuación para disoluciones de no
electrolitos:
,
y definido como el cociente entre el valor experimental de la presión osmótica
media y el valor teórico que se deduce con la ecuación para disoluciones de no
electrolitos:



Disoluciones reales
Para disoluciones concentradas, se ha de utilizar la actividad (más concretamente su logaritmo, )
en lugar de las fracciones molares, y se debe tener en
cuenta la influencia de la presión en el volumen molar del disolvente (en la deducción
termodinámica se considera el volumen de disolvente constante). De esta manera
se obtiene una expresión más precisa, aplicable a las disoluciones concentradas
que contienen un término añadido donde aparece la presión osmótica al
cuadrado:
)
en lugar de las fracciones molares, y se debe tener en
cuenta la influencia de la presión en el volumen molar del disolvente (en la deducción
termodinámica se considera el volumen de disolvente constante). De esta manera
se obtiene una expresión más precisa, aplicable a las disoluciones concentradas
que contienen un término añadido donde aparece la presión osmótica al
cuadrado:

 es el coeficiente
de compresibilidad isotermo del disolvente.[14]
es el coeficiente
de compresibilidad isotermo del disolvente.[14]
En el caso de electrolitos, el coeficiente de actividad del disolvente permanece cercano a la unidad, aunque la concentración de electrolitos sea alta, y por tanto, con coeficientes de actividad del electrolito alejados de la unidad. Por este motivo, el coeficiente de actividad del disolvente no resulta adecuado para caracterizar las propiedades reales de la disolución electrolítica concentrada y se define el coeficiente osmótico (ϕx), por parte del químico danés Niels J. Bjerrum, de la siguiente manera:[mostrar]Deducción termodinámica de la ley de la presión osmótica para disoluciones reales[14]


La presión osmótica en la naturaleza
La presión osmótica en el medio interno

Difusión de agua en las células vegetales por efecto de la presión osmótica.La ósmosis tiene una gran importancia en los seres vivos. Las células de los organismos están rodeadas por fluidos acuosos, como la sangre, la linfa, o la savia, que contienen concentraciones de diferentes solutos. Las membranas celulares son permeables al agua, al oxígeno, al nitrógeno, al dióxido de carbono, y a otras moléculas orgánicas de pequeño tamaño, como glucosa o aminoácidos, mientras que son impermeables a las moléculas poliméricas, como proteínas y polisacáridos. En cambio, los iones inorgánicos y los disacáridos, como la sacarosa, pasan muy lentamente a través de las membranas celulares.
Las células también tienen la capacidad de transportar especies químicas a través de su membrana desde una región de baja concentración de la especie a una región de concentración más elevada, en sentido contrario al del flujo espontáneo. Los mecanismos que originan este tipo de transporte, denominado transporte activo, son complejos y todavía no se conocen totalmente. Un ejemplo típico de transporte activo es el de cationes potasio, K+, hacia el interior de las células desde los líquidos circundantes, que tienen menor concentración de cationes potasio.
En ausencia de transporte activo, la membrana celular permite el paso de moléculas de agua y de todos los solutos permeables hasta que se igualen sus respectivos potenciales químicos a ambos lados de la membrana. No obstante, existe un gran número de especies, tanto en el fluido que rodea la célula como en el fluido celular o citoplasma, que no pueden atravesar la membrana. Si la concentración total de este soluto es más grande en el fluido que rodea la célula, esta perderá agua por ósmosis, y se dice que el fluido circundante es hipertónico respecto al fluido celular (tiene mayor presión osmótica). En caso contrario, cuando la concentración total del soluto que no puede atravesar la membrana es mayor en el fluido de la célula, esta ganará agua del líquido hipotónico circundante (de menor presión osmótica). Cuando no se produce transferencia neta de agua entre el fluido celular y el que rodea la célula, se dice que los dos fluidos son isotónicos, es decir, tienen la misma presión osmótica. La sangre y la linfa son aproximadamente isotónicos respecto de las células de un organismo.
Los líquidos de las inyecciones contienen una disolución salina isotónica con la sangre, porque si se inyectara agua directamente, los eritrocitos de la sangre la absorberían por ósmosis hasta estallar.[7]
Osmorreceptores
Existen unas células especializadas en detectar cambios de la presión osmótica en los líquidos corporales (plasma sanguíneo, fluido intercelular...), llamadas osmorreceptores. Los osmorreceptores pueden localizarse en algunas estructuras, como en el hipotálamo, en las arterias, entre otros lugares. Mediante estas células, el organismo puede regular la concentración de sales en sus fluidos, la osmolalidad.[16] En los seres humanos, son osmorreceptores los núcleos supraóptico o paraventricular. Por medio de las hormonas antidiuréticas (también llamada vasopresina, ADH o pitresina), los osmorreceptores inhiben la diuresis con tal de mantener la presión osmótica de la sangre correspondiente a una osmolalidad media de 285 osmol/kg. Es suficiente una variación de tan sólo 3 osmol/kg para activar este mecanismo.[17]
La absorción de agua en los vegetales
Los vegetales utilizan la presión osmótica para hacer ascender agua a través del xilema desde las raíces hasta las hojas. Así se ha comprobado en los arces al comenzar la primavera y se inicia el movimiento de la savia. Las raíces tienen una gran concentración de azúcares almacenados, producidos durante el verano. Cuando se funde la nieve, el agua llega en grandes cantidades a las raíces y entra dentro de ellas a través de pelos absorbentes, y por efecto de la presión osmótica la savia sube hacia las hojas. Sin embargo, en la mayoría de los vegetales no es posible explicar la ascensión de la savia hasta las hojas por medio de la presión osmótica. En las partes más elevadas del vegetal, la savia asciende debido al descenso de presión provocado por la transpiración de las hojas. En ocasiones, en lugar de que las raíces presenten grandes concentraciones de azúcares, se encuentran rodeadas de agua muy salada, produciéndose el fenómeno conocido como ósmosis inversa; este es el caso de los mangles.[18]
Semillas germinadas con la radícula llena de pelos absorbentes.
Organismos osmófilos
Se denominan osmófilos los organismos extremófilos que pueden crecer en ambientes con una alta concentración de azúcares.[16] Casi todos los microorganismos osmófilos son levaduras. Los osmófilos son similares a los organismos halófilos (medios salinos). La alta concentración de azúcar representa un factor limitante en el crecimiento de muchos microorganismos, pero los osmófilos logran protegerse contra la elevada presión osmótica por medio de la síntesis de osmoprotectores, como alcoholes y aminoácidos. Estos organismos son importantes porque causan el deterioro de productos como los zumos de frutas, los jarabes o la miel, además de tener aplicaciones industriales como la fermentación de la cerveza. Algunos ejemplos de este tipo de microorganismos osmófilos son: Saccharomyces rouxii, Saccharomyces bailii, Debaryomyces, y Saccharomyces cerevisiae, la levadura de la cerveza. "Saccharomyces" deriva del griego, que significa "moho del azúcar".
Aplicaciones
Científicas
Una de las aplicaciones científicas de la presión osmótica es la determinación de masas moleculares o masas molares, de macromoléculas. A partir de la ecuación de van 't Hoff se puede despejar la masa molar, M, del soluto disuelto:


Analíticas
En los laboratorios se puede determinar la concentración total de partículas en disolución osmóticamente activas, lo que se conoce como osmolaridad u osmolalidad, midiendo la presión osmótica. Los aparatos que se utilizan se denominan osmómetros de membrana. Se trata de aparatos automatizados que permiten la realización de las determinaciones de presión osmótica de forma rápida.











 ),
podemos desarrollar
),
podemos desarrollar 




 el volumen de disolvente.
el volumen de disolvente.
 (c es la concentración molar de la disolución). De este modo se obtiene
la ecuación de van 't Hoff:
(c es la concentración molar de la disolución). De este modo se obtiene
la ecuación de van 't Hoff:









EJERCICIOS DE PRESION
OSMÓTICA
- http://www.youtube.com/watch?v=GKR87fvfKSA
- http://servicios.encb.ipn.mx/polilibros/fisicoquimica/PRESION%20DE%20VAPOR/problemas.htm
- http://fluidos.eia.edu.co/hidraulica/articuloses/flujodegases/presiondevapor/presiondevapor.html
- REID, Robert C y SHERWOOD, Thomas K. propiedades de los gases y líquidos. Unión tipográfica editorial hispano-americana. México.1968
- http://es.wikipedia.org/wiki/Presi%C3%B3n_de_vapor
No hay comentarios:
Publicar un comentario